Atossa Therapeutics has received Rare Pediatric Disease (RPD) designation from the U.S. Food and Drug Administration for (Z)-Endoxifen in Duchenne muscular dystrophy (DMD), a strategic shift that positions the estrogen receptor modulator for possible use in pediatric neuromuscular disorders. If approved, the program could qualify for a transferable Priority Review Voucher, but the timeline and regulatory window remain uncertain.
What this reveals about Atossa’s shift toward platform versatility
The decision to pursue a rare disease designation for (Z)-Endoxifen indicates a deliberate broadening of Atossa Therapeutics’ clinical strategy beyond its traditional oncology pipeline. The Seattle-based clinical-stage biopharmaceutical company has spent the past several years positioning (Z)-Endoxifen as a differentiated selective estrogen receptor modulator and degrader for hormone-driven cancers, especially breast cancer. Its push into DMD reframes the compound as a potential platform molecule, capable of impacting estrogen receptor pathways and related kinase activity in non-oncologic tissues.
This move also highlights an emerging industry trend where known molecules with established safety profiles in one indication are being repurposed into orphan diseases with high unmet need. While the molecular rationale for estrogen receptor targeting in muscle pathophysiology remains underexplored in clinical settings, Atossa’s decision reflects a willingness to capitalize on mechanistic hypotheses that may apply across therapeutic areas. The company has not yet released human data on (Z)-Endoxifen in DMD, but its regulatory bet signals growing confidence in preclinical insights and the broader role of hormonal signaling modulation in pediatric muscle degeneration.
Why the priority review voucher pathway is both a financial and strategic hedge
Atossa’s announcement explicitly ties the RPD designation to the potential receipt of a Priority Review Voucher (PRV), which can either be used to expedite a future New Drug Application or sold to another sponsor. These vouchers remain among the most coveted assets in early-stage biopharma, with transactions between 2023 and 2025 ranging from $100 million to $160 million per voucher, depending on market conditions and urgency from Big Pharma buyers.
Yet the PRV program itself faces regulatory uncertainty. Under current rules, only drug programs designated as rare pediatric diseases before December 20, 2024, and approved before September 30, 2026, are eligible. Atossa’s window for both designation and potential approval now sits in a narrow and politically sensitive zone. A legislative proposal titled the “Give Kids a Chance Act” has passed the U.S. House of Representatives and aims to extend the program through 2029, with retroactive eligibility, but the bill remains pending in the Senate.
Analysts tracking the regulatory space believe that if the bill is not passed by mid-2026, many smaller biotech firms banking on PRVs for funding cycles may face valuation resets. Atossa’s position is relatively early, so even a slight delay in program advancement could jeopardize voucher eligibility. For now, the RPD designation gives the company access to enhanced FDA guidance on trial design and regulatory expectations, but the long-term financial value will depend on timing and execution.
How (Z)-Endoxifen differs from exon-skipping and gene therapy approaches in DMD
The DMD space is dominated by exon-skipping therapies like eteplirsen (Exondys 51), as well as emerging gene therapies targeting dystrophin replacement. Most of these treatments are mutation-specific, applying only to subsets of the DMD patient population with particular exon deletions. By contrast, Atossa’s (Z)-Endoxifen, a SERM/D, does not require mutation stratification and may theoretically address a broader patient population.
According to statements from the company’s R&D leadership, (Z)-Endoxifen’s potential benefit in DMD could stem from its non-exon-specific mechanisms and possible downstream modulation of muscle repair and inflammatory pathways. The molecule’s dual activity—including PKC inhibition—suggests a potential for modulating intracellular signaling networks involved in muscle degeneration, fibrosis, and possibly regeneration. Still, this hypothesis remains speculative without human clinical data or validated preclinical models in dystrophin-deficient systems.
Unlike gene therapy programs that target dystrophin restoration and are often limited by immunogenicity, viral vector delivery challenges, or high manufacturing costs, a small molecule like (Z)-Endoxifen offers a different value proposition. If effective, it could represent an orally administered, broadly accessible therapeutic approach. However, the complete absence of safety or efficacy data in pediatric neuromuscular populations introduces major translational risk.
Why trial design will define the credibility of Atossa’s expansion into DMD
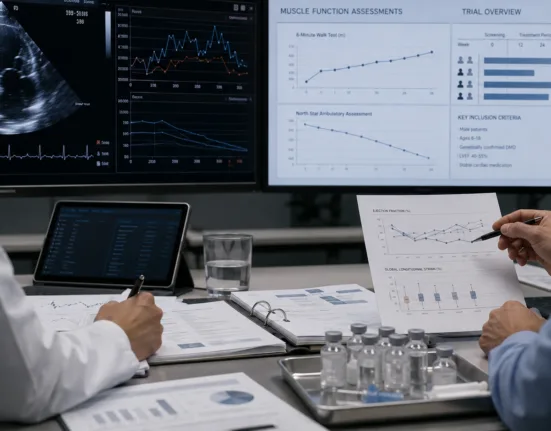
For Atossa Therapeutics to be taken seriously in the DMD space, it will need to articulate a clinical strategy that clearly delineates mechanism of action, outcome measures, and patient eligibility. Unlike oncology, where biomarkers such as ER status or progression-free survival are well-established, neuromuscular trials demand functional endpoints such as six-minute walk tests, North Star Ambulatory Assessment scores, or forced vital capacity in declining adolescents.
Moreover, the pediatric context imposes additional layers of complexity. Regulatory watchers suggest that any proposed trial involving hormonal modulation in boys with DMD will face intense scrutiny over long-term safety, endocrine disruption, and off-target effects. Industry observers also note that placebo-controlled trials in DMD are ethically sensitive, with increasing expectations for external control arms, digital monitoring tools, and natural history data integration.
Atossa has not yet disclosed its intended trial design, enrollment timeline, or site selection strategy. The absence of an active IND for DMD-specific trials suggests that the designation is still in a preclinical or regulatory planning phase. While the company has referenced past experience in obtaining IND clearance for DMD-related programs, including eteplirsen, it is unclear whether those capabilities have translated into tangible infrastructure for this new indication.
What the intellectual property strategy implies about long-term intent
One of the less discussed yet strategically significant aspects of this announcement is the reinforcement of Atossa’s intellectual property posture. The company has previously disclosed a growing global patent estate for (Z)-Endoxifen, including recently issued U.S. patents and a slate of pending filings worldwide. Expanding the scope of this estate to include rare pediatric neuromuscular claims could serve multiple purposes.
First, it adds potential value to licensing discussions. Even if Atossa does not fully develop (Z)-Endoxifen in DMD, patent coverage in this area could make the program attractive to neuromuscular-focused biotechs or academic spinouts looking for small-molecule alternatives to biologics. Second, it may insulate the company from future challenges related to obviousness or prior art, especially if the estrogen receptor or PKC pathways become more validated in muscle disease over time.
Third, it could boost Atossa’s attractiveness to acquirers seeking early-stage, non-dilutive PRV pathways. Companies looking to gain FDA priority reviews for other pipeline products may view Atossa’s RPD designation as a buyable asset, particularly if the PRV extension bill is passed and (Z)-Endoxifen clears IND and Phase 1 milestones.
Why clinicians and payors will demand clearer mechanistic rationale
Despite the regulatory enthusiasm, clinical uptake of any future (Z)-Endoxifen DMD therapy would depend heavily on its mechanistic clarity and differentiation. In oncology, SERMs are widely understood and used, but their role in muscle biology is less intuitive. Endocrinologists and neuromuscular specialists are likely to demand clear evidence that the molecule produces functionally meaningful improvements in muscle strength, disease progression, or respiratory outcomes.
Reimbursement bodies will also require cost-effectiveness data, especially given the recent controversies around the pricing of gene therapies and high-cost exon-skipping drugs. A small-molecule alternative could be more affordable and scalable, but only if it delivers durable benefit across broad patient groups. Without early biomarker data or predictive signals, Atossa may face challenges in shaping payer and clinician expectations.
What could go wrong next for the program
The largest risk is translational: moving a hormone-modulating oncology drug into a pediatric neuromuscular population carries both scientific and regulatory uncertainty. The pathway from preclinical promise to clinical viability in DMD is notoriously challenging, with high attrition rates and frequent failures even among well-capitalized developers.
Political risk is also real. If the “Give Kids a Chance Act” fails in the Senate or stalls indefinitely, Atossa’s RPD designation may not lead to a voucher. That would erode a key pillar of the business case for investing in this indication, particularly if internal resources are diverted from ongoing oncology programs.
Finally, timing will be critical. Investors and partners will expect visible progress in the form of IND submission, site activation, or early patient enrollment in 2026. Any material delay could diminish the perceived value of the designation and reduce Atossa’s optionality in licensing or capital markets.