Endospan has received United States Food and Drug Administration approval for the NEXUS Aortic Arch Stent Graft System, a bimodular endovascular device intended for minimally invasive treatment of aortic arch disease, including chronic aortic dissections, in patients considered high risk for conventional open surgical repair. The approval appears to be a premarket approval decision backed by one-year data from the TRIOMPHE investigational device exemption study, clearing the way for a United States commercial launch in one of the most technically difficult segments of aortic intervention.
Why FDA approval of NEXUS matters far beyond a single device launch in a narrow specialty
The real significance of this approval is that it targets a part of the aorta where treatment has long lagged behind the rest of endovascular repair. Thoracic endovascular aortic repair is well established in other segments, but the arch remains a procedural headache because of tortuous anatomy, supra-aortic branch involvement, embolic risk, and the still-serious danger of stroke or retrograde complications. Recent reviews note that few fully endovascular arch options have been available in the United States, which is why open surgery or hybrid debranching has remained the default despite the burden that approach places on frail patients.
That is why NEXUS is more than just another stent graft iteration. What is genuinely new here is not the broad idea of arch endografting, which has been in development for years, but the regulatory validation of an off-the-shelf branched system designed specifically for the ascending aorta and arch. Endospan says the platform uses a bimodular configuration with an integrated brachiocephalic trunk branch, a pre-curved ascending component, and a 20F delivery system intended to reduce arch manipulation. Those design claims matter because arch repair has always been a mechanics problem before it becomes a market problem.
How the TRIOMPHE data strengthens the case while still leaving important evidence gaps unresolved
The pivotal support looks directionally encouraging, especially given the risk profile of the enrolled population. External reporting tied to the approval states that the TRIOMPHE IDE trial showed about 90% survival from lesion-related death at one year, roughly 90% freedom from disabling stroke, and about 98% freedom from reintervention due to endoleaks in a high-risk population. Earlier reporting around the same program described TRIOMPHE as a prospective, multicenter, non-randomized, three-arm study conducted at 30 U.S. aortic centers and one in New Zealand, with chronic dissection, aneurysm, and related arch pathologies represented in the broader study design.
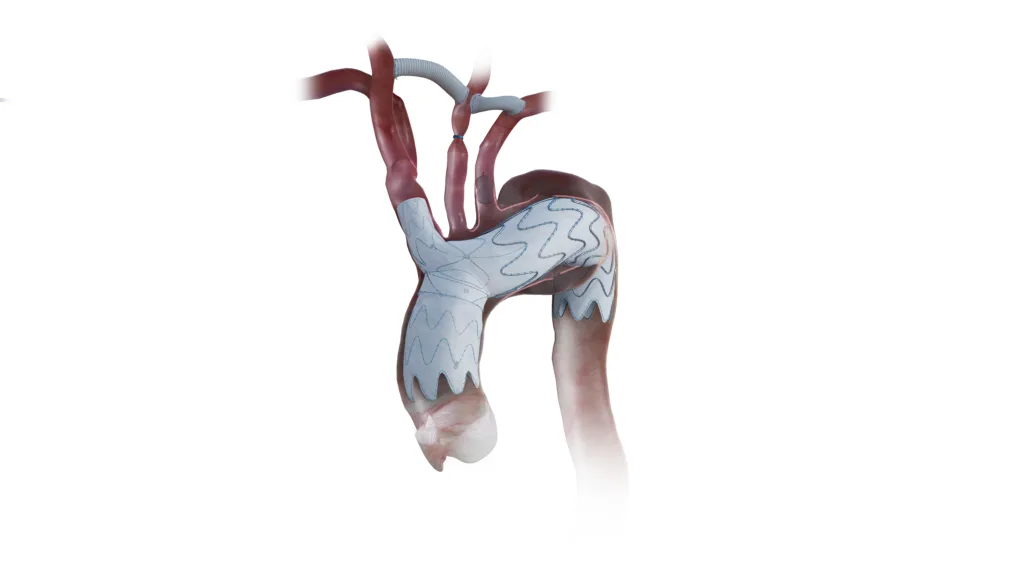
Still, clinicians and regulators will read these results with appropriate caution. The study was prospective and multicenter, which adds weight, but it was not randomized, and the most persuasive comparator for aortic arch treatment remains difficult to establish because open surgery candidates and high-risk endovascular candidates are not interchangeable groups. That means NEXUS is entering practice on the strength of credible but still selective evidence rather than on a head-to-head proof of superiority over open or hybrid arch repair. In this field, that is not unusual, but it does limit how aggressively adoption claims should be interpreted.
The other nuance is that one-year follow-up is necessary but not sufficient in arch devices. Durability, branch patency, device migration, endoleak behavior, and late neurologic events matter enormously in this anatomy. Published and conference-linked NEXUS experience from Europe has been promising, including multicenter reports and observational follow-up, but those datasets do not erase the need for longer-term U.S. surveillance in a post-approval setting. In aortic repair, the first year tells you whether a platform can work. Years three to five tell you whether it can stay put, stay patent, and stay commercially credible.
What this approval could change for surgeons treating chronic dissection and complex arch anatomy
For surgeons and interventional teams, the practical appeal is clear. The hardest patients in arch disease are often the ones least able to tolerate sternotomy, circulatory arrest, or complex redo procedures. A less invasive option for chronic dissection and selected arch lesions could expand the treatable pool, especially where open risk has been prohibitive. Endospan has argued that a large share of thoracic arch disease remains undiagnosed or untreated, and while such market sizing should be treated cautiously, the underlying clinical point is credible: many patients sit in a grey zone between anatomical complexity and surgical unsuitability.
But the approval does not magically eliminate the procedural ecosystem required to use the device well. Arch endovascular repair still depends on careful imaging, landing-zone assessment, cerebral protection strategy, branch management, and center experience. Some NEXUS-related reports have discussed staged cervical debranching or adjunctive revascularization strategies, underscoring that this is not a plug-and-play peripheral intervention. In other words, the device may broaden options, but it will likely remain concentrated in experienced aortic centers rather than dispersing rapidly across all vascular programs.
That center-of-excellence dynamic matters commercially too. Devices that address small but high-acuity populations can still become strategically valuable if they deepen relationships with referral hospitals and create a defensible niche. Yet scalability in arch repair is never just about regulatory clearance. It is about training, case selection, reimbursement comfort, multidisciplinary buy-in, and whether early adopters can reproduce the stroke and mortality profile seen in trials once the device meets broader real-world anatomy. The arch is where optimism goes to get audited by workflow.
Why the FDA nod also has strategic importance for Artivion and the broader aortic device market
The approval also matters because of who is standing behind it. Artivion, Endospan’s long-standing partner, said the FDA approval gives it the right to exercise an option to acquire Endospan within 90 days and that it has financing lined up in anticipation of that possibility. That turns a regulatory event into a strategic corporate trigger. If Artivion proceeds, NEXUS could become part of a broader aortic portfolio rather than a standalone specialty asset, which may improve commercialization discipline and physician reach in the United States market.
Investor sentiment appears to be reading the news as incrementally positive. Artivion shares were trading at $35.22 on April 7, 2026, up about 5.4% on the day, suggesting the market viewed the approval and acquisition optionality constructively, even if the company still carries execution questions and negative trailing earnings. For investors, the next issue is less whether the approval matters and more whether NEXUS can become a meaningful contributor without diluting focus or pressuring integration economics if Artivion exercises the option.
What clinicians, regulators, and industry watchers are likely to watch next after the NEXUS launch
The next watchpoints are straightforward. Clinicians will want clearer subgroup visibility, especially around chronic dissection versus aneurysm, neurologic outcomes, reintervention patterns, and how performance varies with adjunctive bypass or debranching. Regulators and hospital value committees will be watching whether real-world results preserve the favorable one-year safety signals. Industry observers will also want to know how often patients initially screened for NEXUS are ultimately anatomically eligible, because arch devices can look more scalable on slides than they do on preoperative imaging workstations.
The bigger picture is that NEXUS does not end the debate over how best to treat aortic arch disease. What it does is move the conversation from theoretical feasibility to approved clinical availability in the United States. That is a meaningful shift for a field where the arch has remained the last really stubborn frontier of endovascular aortic therapy. The approval gives high-risk patients and specialist centers a new tool, but the next chapter will be decided by durability, reproducibility, and whether post-approval practice confirms that the promise of minimally invasive arch repair can survive contact with real-world anatomy.