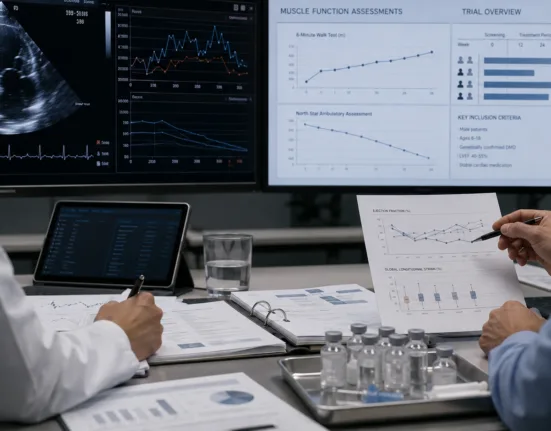
Sarepta Therapeutics has announced positive three-year functional data from Part 1 of its global Phase 3 EMBARK study of ELEVIDYS (delandistrogene moxeparvovec-rokl), the only approved gene therapy for Duchenne muscular dystrophy (DMD). In boys aged four to seven at baseline, the gene therapy showed statistically significant and clinically meaningful slowing of disease progression on key functional endpoints, including NSAA, Time to Rise, and 10-meter walk/run, compared to a propensity-weighted external control. No new safety signals emerged.
What this confirms about ELEVIDYS as a disease-modifying therapy in Duchenne
The updated EMBARK readout is the first long-term, controlled dataset to demonstrate that ELEVIDYS not only maintains but strengthens functional benefits in Duchenne patients over time. While previous data spanning up to five years existed in smaller cohorts, the new three-year results from 52 children treated in Part 1 of the trial validate sustained motor improvements and a widening gap versus the natural history of untreated DMD.
By year three, ELEVIDYS-treated patients showed a +4.39-point improvement on the North Star Ambulatory Assessment, compared to baseline, while the external control group declined. Notably, Time to Rise improved by 6.05 seconds and 10-meter walk/run improved by 2.70 seconds relative to controls, translating to a 73% and 70% slowing of disease progression, respectively.
From a clinical standpoint, these outcomes challenge the long-held assumption that gene therapies in Duchenne may offer transient benefit without altering the underlying trajectory. ELEVIDYS appears to shift the disease course—at least in the ambulatory population—toward preservation of function well past the expected onset of irreversible decline, typically seen around 9–10 years of age.
Industry observers believe this is the most compelling evidence to date that AAV-based gene therapy can act as a true disease-modifying agent in neuromuscular disorders. While not curative, the durability signal over a 3-year span represents a substantial clinical milestone.
Why EMBARK matters for setting new regulatory and reimbursement precedents
From a regulatory standpoint, the ELEVIDYS dataset introduces fresh considerations for long-term evidence requirements in rare disease gene therapy approvals. Sarepta’s ability to maintain a positive delta over external controls across three years strengthens the FDA’s accelerated approval decision and reduces the likelihood of label narrowing. However, the absence of a placebo comparator beyond the first year introduces limitations that some regulators outside the U.S. may scrutinize.
Payers and health technology assessment (HTA) bodies—especially in Europe—are likely to weigh these results against high upfront costs and real-world effectiveness in broader populations. While the magnitude of effect on TTR and 10MWR is clear, whether these translate into functional independence or delayed loss of ambulation remains to be proven in registries.
Clinicians tracking the field note that Duchenne’s heterogeneity and the role of corticosteroids complicate cross-trial comparisons, but the external control methodology here—anchored in three separate datasets with matched covariates—has become a pragmatic standard for long-term studies in rare diseases where placebo control is infeasible.
What this changes in physician confidence and standard-of-care transition timing
The durability data may accelerate a shift in when and how Duchenne gene therapy is deployed in standard clinical practice. Historically, clinicians have been cautious in treating children close to the lower end of the eligibility window, wary of overpromising outcomes. However, the three-year data show that initiating therapy between ages 4 and 7 may provide the highest probability of durable benefit.
This could sharpen the focus on early genetic screening and lead to changes in neuromuscular practice patterns, particularly in countries where newborn screening for DMD is already under consideration. Some pediatric neurologists suggest that long-term confirmation of ELEVIDYS’s benefit at the 9–10 year age mark could shift patient expectations and physician counseling around therapy timing.
Still, the modest size of the cohort (n=52) and the continued need for corticosteroid use alongside gene therapy remain operational constraints in real-world use. Also, the EMBARK dataset does not yet include non-ambulatory populations, where safety risks have been more pronounced.
What this reveals about gene therapy durability and ELEVIDYS’s underlying platform
Beyond functional endpoints, the EMBARK dataset reinforces the mechanistic plausibility of micro-dystrophin gene transfer using AAVrh74. Although data on dystrophin expression in the three-year timeframe were not presented, prior biomarker results in the EMBARK subset and other studies support consistent transgene expression up to 12 weeks post-infusion.
This durability of effect suggests that once transduced, skeletal muscle cells can sustain therapeutic benefit despite turnover and immune clearance pressures. However, longer-term questions persist around whether benefits plateau or decline in later adolescence when muscle degeneration accelerates.
Preclinical and small-scale follow-up studies hint at a ceiling effect for micro-dystrophin therapies beyond age 12, but the current readout reinforces the case for early administration as the best strategy to lock in functional gains.
Analysts also point to this as a milestone for AAV-based platforms more broadly, especially as other gene therapies in neuromuscular and ophthalmic diseases seek to demonstrate multi-year durability.
What risks remain from a safety and real-world implementation perspective
While no new treatment-related safety signals were observed, the safety profile of ELEVIDYS is not without concern. Acute liver injury, thrombocytopenia, troponin-I elevations, and rare immune-mediated myositis remain part of the risk landscape, especially in older or non-ambulatory patients.
The boxed warning around acute serious liver injury remains a major clinical consideration. Fatal cases have been documented in non-ambulatory populations, leading to more stringent monitoring protocols and extended corticosteroid regimens. Immunogenicity-related concerns, including myocarditis and preexisting AAVrh74 antibodies, also remain gating factors for some patients.
Sarepta’s updated FDA label now limits use to ambulatory individuals aged 4 and older with a confirmed DMD gene mutation and excludes certain exon deletions (exon 8 and/or 9). These restrictions may complicate physician decision-making, especially in mixed-mutation families or resource-limited geographies where genomic testing is not always timely.
Real-world data collection will be critical in verifying whether the EMBARK durability results translate across patient subtypes and healthcare settings. Expanded follow-up from the EXPEDITION study, ongoing long-term registries, and broader surveillance through commercial rollout will need to fill in these gaps.
What this unlocks for Sarepta’s global strategy with Roche and Chugai
With more than 1,200 patients dosed globally and new evidence of sustained benefit, Sarepta and its partners—Roche for ex-U.S. markets and Chugai in Japan—are likely to push harder on regulatory and commercial expansion.
These results could facilitate label broadening in markets where regulatory authorities are seeking robust functional data before approval. In the U.S., they reinforce Sarepta’s role as a precision genetic medicine leader and strengthen its pipeline credibility as additional neuromuscular and limb-girdle programs advance.
However, the commercial challenge remains significant. Manufacturing scalability, AAV vector supply chain logistics, and site-of-care readiness are all gating factors to broader adoption, especially as insurers and payers demand real-world confirmation of cost-effectiveness.
What payers, clinicians, and regulators are likely to monitor next
Several key areas will dominate the next phase of scrutiny around ELEVIDYS. First is long-term data beyond three years, particularly as treated boys enter adolescence and muscle mass begins to decline more rapidly.
Second, payers will likely seek head-to-head comparisons or real-world benchmarks to understand the value proposition versus corticosteroids, exon-skipping therapies, and future CRISPR-based interventions.
Third, regulators may ask for more clarity on re-dosing potential, given the immunogenicity of AAV vectors, and explore new vector engineering strategies to support retreatment if functional decline resumes after 5–7 years.
Finally, clinicians will monitor quality-of-life data, particularly on patient-reported outcomes, upper limb function, and preservation of independence, to understand whether ELEVIDYS meaningfully extends not just mobility, but daily life participation.