CGBIO has received U.S. Food and Drug Administration 510(k) clearance for EASYMADE TI, its patient-specific titanium implant for cranial and non-load-bearing craniofacial reconstruction. The clearance gives the South Korean medical device manufacturer a regulatory route into the U.S. market at a time when customized reconstruction, 3D-printed titanium implants, and digitally planned surgical devices are moving from specialist use cases toward more scalable hospital workflows.
The clearance is more than a narrow device milestone because it places CGBIO inside one of the more technically demanding corners of personalized medtech. Cranial and craniofacial reconstruction requires a device that is not merely biocompatible and structurally reliable, but also anatomically matched to the individual patient’s defect. That distinction matters because patient-specific implants sit at the intersection of imaging, design software, advanced manufacturing, surgeon workflow, sterilization logistics, and regulatory documentation. A clearance in this category therefore signals not only that EASYMADE TI met the substantial equivalence threshold under the FDA 510(k) pathway, but also that CGBIO has built a process that can be translated into the highly regulated U.S. device market.
Why CGBIO’s EASYMADE TI clearance could matter for patient-specific cranial implants in the United States
The most immediate significance is that EASYMADE TI is intended for a reconstruction category where standard implants can be clinically workable but surgically imperfect. Cranial and craniofacial defects often follow trauma, tumor removal, decompressive craniectomy, infection, or prior surgical intervention. In such cases, surgeons may need to restore protective coverage, symmetry, contour, and functional anatomy while reducing intraoperative adjustment. A patient-specific titanium implant can address those needs by being designed from computed tomography data before the patient reaches the operating room.
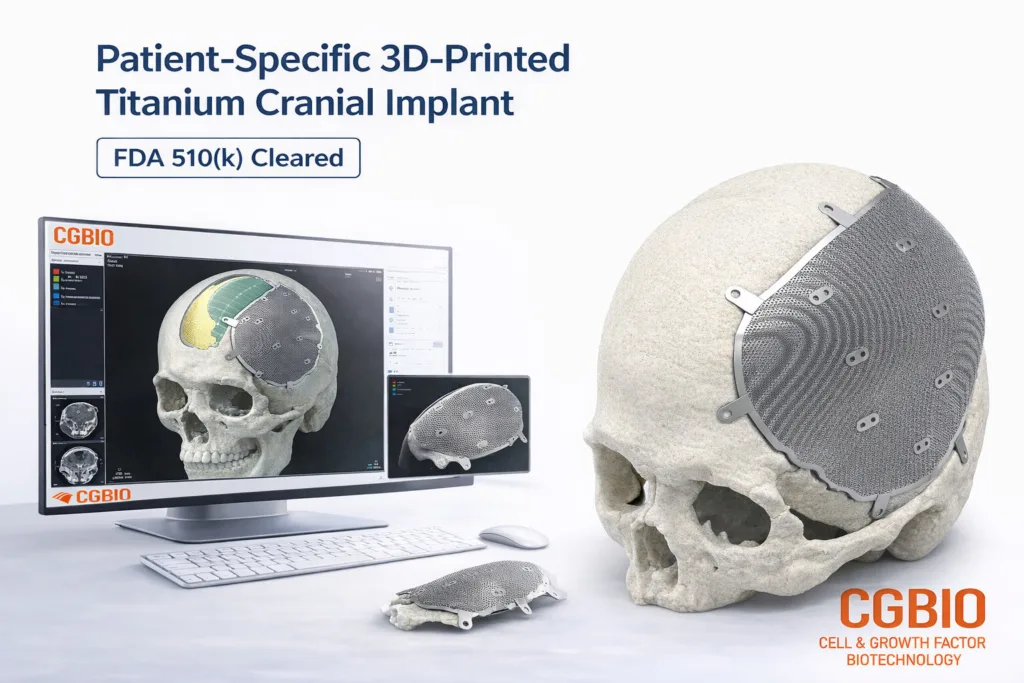
That does not automatically make patient-specific implants superior in every case. Surgeons still weigh defect size, location, soft-tissue condition, infection risk, patient history, operating time, cost, hospital procurement processes, and the availability of alternatives such as polymethyl methacrylate, polyetheretherketone, autologous bone, or other titanium mesh products. The clearance therefore gives CGBIO market access, not guaranteed adoption. Its commercial test will be whether U.S. surgeons and hospitals see enough procedural value in EASYMADE TI to justify adding a new overseas design and manufacturing workflow to their existing reconstruction pathways.
The more interesting commercial question is whether CGBIO can turn personalization into a repeatable supply model. Patient-specific devices have often been attractive in concept but operationally complicated in practice. Imaging quality must be sufficient, segmentation and design must be accurate, surgeon review must be efficient, production must be reliable, and the final implant must arrive on a timetable that fits surgical scheduling. CGBIO’s stated five-day delivery process will therefore be watched closely because speed is not a marketing detail in this category. It may determine whether the device is practical for routine reconstruction planning or remains confined to selected cases.
How the FDA 510(k) pathway shapes the commercial opportunity and the limits of the claim
The FDA 510(k) clearance route is important because it is based on substantial equivalence to a legally marketed predicate device rather than the kind of de novo risk evaluation or premarket approval pathway used for higher-risk or first-of-kind products. For CGBIO, that provides a clearer route to U.S. commercialization, but it also means the clearance should not be read as clinical proof that EASYMADE TI delivers better outcomes than every competing implant option. The regulatory achievement validates market entry under the 510(k) framework. It does not settle questions around comparative surgical performance, long-term revision rates, reimbursement dynamics, or surgeon preference.
That distinction matters for clinicians and procurement teams. A cleared patient-specific implant can enter hospital evaluation, but adoption still depends on evidence, experience, service reliability, and economics. Hospitals may ask whether CGBIO can provide responsive design revisions, technical support across time zones, documentation for operating room teams, and consistency across different U.S. healthcare systems. In craniofacial reconstruction, the device is only one part of the value chain. The service model around the device can be just as important as the implant itself.
The clearance also gives CGBIO a regulatory reference point for future expansion. If EASYMADE TI establishes credibility in the United States, it may strengthen the South Korean medical device manufacturer’s ability to position additional patient-specific reconstruction products in global markets. However, each product, indication, material, or manufacturing variation can carry its own regulatory and quality-system demands. A single clearance is a foundation, not a passport for an entire pipeline.
What EASYMADE TI reveals about the role of 3D-printed titanium in reconstructive surgery
EASYMADE TI is manufactured from medical-grade titanium alloy using Laser Powder Bed Fusion, a metal 3D printing process that can produce complex geometries and patient-matched forms. This is clinically relevant because cranial and craniofacial implants often require curved surfaces, contour fidelity, edge accuracy, and structural stability. Additive manufacturing can reduce the constraints associated with traditional machining or intraoperative manual shaping, particularly when the defect geometry is irregular.
The technology angle is compelling, but it brings its own burden. Metal 3D printing in medical devices depends on tight process controls, powder quality management, build validation, post-processing, surface finishing, cleaning, mechanical testing, and traceability. For a patient-specific implant business, manufacturing is not a batch-only operation. Each device has unique geometry, which places heavy emphasis on validated workflows rather than simple mass production. This makes quality systems, documentation, and process repeatability critical to commercial credibility.
That is where CGBIO’s model will face its most practical test. Designing in Korea and delivering to U.S. hospitals within five days could be attractive, especially for scheduled reconstruction procedures. However, cross-border logistics, case review timelines, customs handling, hospital sterilization readiness, and surgeon-specific design preferences can introduce friction. In personalized medtech, the promise is precision. The risk is that operational complexity can quietly erode the advantage if not managed tightly.
Why U.S. adoption may depend as much on workflow integration as device design
CGBIO’s challenge is not simply to persuade surgeons that a titanium implant can match cranial anatomy. That proposition is already familiar in reconstruction markets. The harder task is to embed EASYMADE TI into the clinical workflow. A U.S. surgeon must be able to submit CT data, review the proposed design, request adjustments where needed, plan the procedure, coordinate sterilization, and trust the final implant to arrive before the scheduled operation. Any delay or design mismatch can disrupt surgical planning and weaken confidence in the platform.
This is why hospital adoption may start with specialist centers, neurosurgical teams, craniofacial surgeons, and institutions that already use patient-specific planning. These centers are more likely to understand the value of customized implants and to tolerate the additional coordination required. Broader adoption may require evidence that EASYMADE TI can reduce operating time, improve fit, support cosmetic reconstruction goals, or lower the burden of intraoperative modification. Without that real-world evidence, the device may compete mainly on service speed, design quality, and surgeon relationships.
Reimbursement will also matter. Even when surgeons like a customized implant, hospitals must assess procurement cost, coding pathways, payer coverage, and overall economic impact. A device that reduces surgical time or revision risk may justify a premium, but that argument must be demonstrated in practice. CGBIO’s U.S. expansion will therefore depend not only on regulatory clearance and technical capability, but also on whether it can build a business case that hospital administrators can defend.
What this clearance changes for Korean medtech companies targeting global device markets
CGBIO’s clearance is strategically notable because it positions a South Korean manufacturer in a specialized U.S. reconstruction segment that requires both technical sophistication and regulatory confidence. South Korea has built strong medical technology and biomanufacturing capabilities, but global device expansion often depends on proving that a company can meet demanding U.S. regulatory, quality, and commercial expectations. EASYMADE TI gives CGBIO a visible case study in that transition.
The broader implication is that Asian medtech companies are no longer competing only through lower-cost manufacturing or regional distribution. More are attempting to enter advanced device categories where software-enabled design, additive manufacturing, biomaterials, and regulatory execution are central to differentiation. In that context, EASYMADE TI is not just a cranial implant. It is a signal that CGBIO wants to compete in the higher-value segment of customized reconstruction.
Still, U.S. medtech expansion is unforgiving. Market access does not equal channel access. CGBIO will need surgeon education, hospital relationships, reimbursement support, quality responsiveness, and probably U.S.-based commercial infrastructure. The first Korean-company distinction may create attention, but long-term success will depend on whether the device becomes trusted in repeat clinical use.
What clinicians, regulators, and industry observers are likely to watch next
The next phase will be defined by commercial execution rather than regulatory announcement value. Clinicians will watch whether EASYMADE TI performs reliably across defect types, whether the fit reduces intraoperative adjustment, and whether the workflow supports practical surgical scheduling. Regulators and quality observers will focus on manufacturing consistency, design-control robustness, and post-market surveillance as the device moves into U.S. use. Industry observers will look for whether CGBIO can convert clearance into recurring hospital adoption or whether the market remains fragmented among established customized implant providers.
For CGBIO, the opportunity is clear but not frictionless. EASYMADE TI enters a field where patient-specific reconstruction is clinically logical, technologically feasible, and increasingly aligned with digital surgery trends. The unresolved question is whether the South Korean medical device manufacturer can turn that logic into scale in the United States. In personalized medtech, the winners are rarely the firms with only the best material or the most advanced printer. They are the firms that make customization feel dependable, fast, and operationally boring for surgeons. That is the real test now facing CGBIO.