Sarepta Therapeutics has confirmed that screening and enrollment are now underway in Cohort 8 of its ENDEAVOR Phase 1b study, marking the company’s most concrete clinical step toward restoring access to its gene therapy delandistrogene moxeparvovec (ELEVIDYS) for non-ambulatory individuals with Duchenne muscular dystrophy. The cohort, cleared by the U.S. Food and Drug Administration for dosing, will evaluate an enhanced immunosuppressive protocol incorporating sirolimus administered before and after gene therapy infusion, in approximately 25 non-ambulatory participants across U.S. trial sites.
Why this cohort exists: the fatal adverse event context that forced Sarepta’s hand
Cohort 8 is not a routine expansion of an existing programme. It is a direct clinical response to a sequence of fatal outcomes in non-ambulatory patients that fundamentally altered the regulatory and commercial status of ELEVIDYS. Following the deaths of at least two, and subsequently three, non-ambulatory patients from acute liver failure after receiving the gene therapy, the FDA required a label update that removed non-ambulatory patients from the approved commercial indication. That exclusion, formalised in November 2025, left a significant and medically vulnerable population without access to the only approved gene therapy for Duchenne. Cohort 8 is the formal mechanism through which Sarepta is attempting to rebuild the evidence base needed to change that.
The clinical rationale for sirolimus as the immunosuppressive agent of choice reflects both preclinical data and a converging regulatory-scientific view that standard corticosteroid-based immunosuppression alone is insufficient for non-ambulatory patients, who appear to mount a more severe hepatic immune response following AAV vector delivery. The FDA itself, according to earlier disclosures, proactively raised the question of whether additional immunosuppression including sirolimus should be considered alongside the gene therapy. That the regulator subsequently cleared Cohort 8 for dosing signals alignment between Sarepta’s proposed protocol and the agency’s assessment of an acceptable investigational risk-benefit framework.
What the sirolimus protocol is actually testing and why the design choices matter
The Cohort 8 regimen involves 14 days of peri-infusion sirolimus dosing beginning before the ELEVIDYS administration, followed by continuation of sirolimus for 12 weeks post-infusion. The dual primary endpoints are the incidence of acute liver injury and dystrophin expression at 12 weeks. That pairing is analytically significant: it positions the cohort to simultaneously address the safety signal that triggered the label restriction and to confirm that the addition of sirolimus does not materially compromise the therapeutic mechanism. If sirolimus suppresses the immune response sufficiently to prevent hepatotoxicity but also blunts the cellular immune engagement required for effective transgene expression, the intervention may trade one problem for another.
Sirolimus, also known as rapamycin, works by inhibiting the mTOR signalling pathway, dampening T-cell proliferation and cytokine production without the broad metabolic consequences associated with prolonged corticosteroid use. Its use as a prophylactic immunosuppressant in AAV gene therapy is not without precedent, and an independent retrospective analysis presented at the World Muscle Society Congress by researchers at Vanderbilt University Medical Center provided early observational support: none of six patients who received sirolimus prior to ELEVIDYS infusion in that series developed acute liver failure. Analysts at William Blair described this as supportive context, characterising the FDA clearance for Cohort 8 as a constructive step toward addressing the safety concerns that have limited the therapy’s reach, and identifying it as a viable pathway toward a broader label. Those observations remain retrospective and derive from a small series, and the Cohort 8 data will need to establish prospective, controlled confirmation before any regulatory submission becomes viable. [SINGLE SOURCE – VERIFY: William Blair analyst commentary cited from Fierce Pharma; confirmation from second independent analyst source not located at time of publication]
The size and design of Cohort 8 raises questions about the evidence threshold ahead
With approximately 25 participants, Cohort 8 is small even by Phase 1b standards. The study design is open-label, which means neither patients nor investigators are blinded to treatment, introducing potential for subjective bias in endpoint assessment. There is no comparator arm. The primary hepatic endpoint relies on the incidence of acute liver injury, a binary safety outcome rather than a graded severity measure, which makes statistical inference on risk reduction inherently limited in a small cohort. Sarepta has not publicly specified the threshold ALI incidence rate it would consider acceptable as evidence of meaningful risk mitigation in the non-ambulatory population.
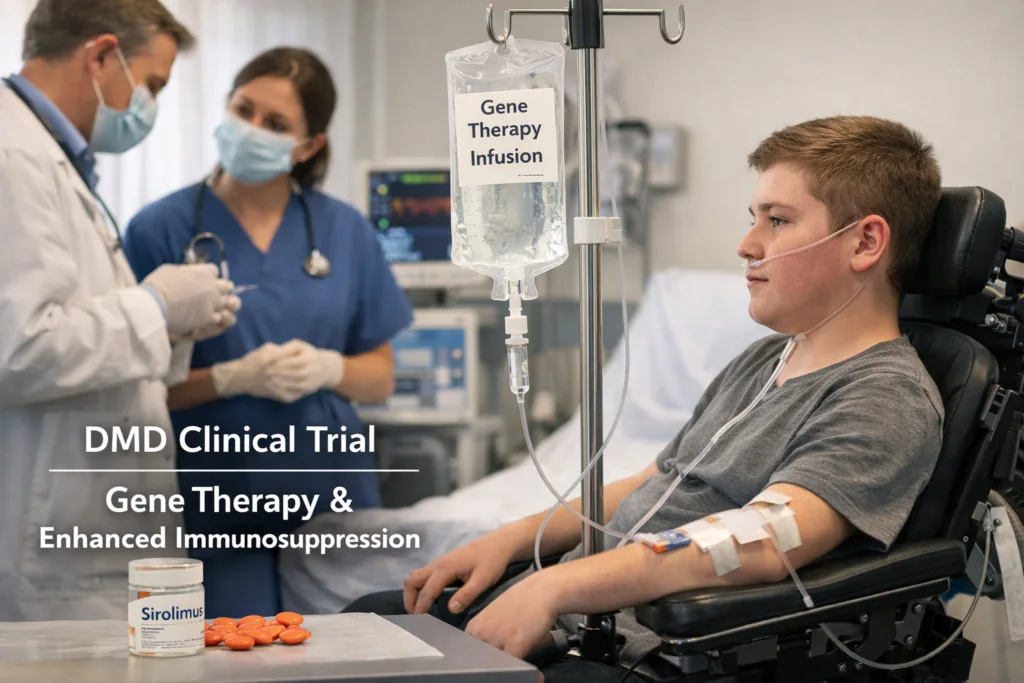
The absence of a placebo or active control also complicates interpretation of the dystrophin expression endpoint. Earlier ENDEAVOR cohorts have generated micro-dystrophin expression data in a range of patient subgroups, but direct comparisons across cohorts are confounded by differences in age, disease stage, ambulatory status, and immunosuppressive background regimens. Whether sirolimus itself affects micro-dystrophin protein quantification by western blot, the primary assay methodology, through any indirect immunological mechanism has not been publicly addressed by the company. These are not disqualifying limitations for a safety-oriented Phase 1b cohort, but they are the questions that will define whether a subsequent regulatory submission for label expansion is achievable or whether the FDA will require additional controlled data.
What label reinstatement for non-ambulatory patients would require from Sarepta
The pathway from Cohort 8 data to a rebroadened label is not automatic. Sarepta will need to demonstrate, to the FDA’s satisfaction, that the sirolimus regimen meaningfully reduces the hepatic risk in non-ambulatory patients without unacceptably compromising efficacy or generating its own serious adverse event profile. The immunosuppression compound itself carries risks including susceptibility to serious infections, a consideration already flagged in ELEVIDYS prescribing information even under a standard corticosteroid regimen. Adding sirolimus for up to 12 weeks in patients with advanced Duchenne, who frequently have compromised respiratory and cardiac function, extends the window of infection vulnerability materially. Serious respiratory infections, including with fatal outcomes, have already been documented in patients on immunosuppressant corticosteroids required for ELEVIDYS administration.
Analysts tracking the file suggest that if Cohort 8 data are favourable, a regulatory submission seeking label expansion for non-ambulatory patients could be submitted as early as 2026, with an FDA decision potentially possible in 2027. That timeline assumes uncomplicated enrolment, no further serious adverse events in the cohort, and a data package the FDA is prepared to act on without requiring longer follow-up or a larger dataset. Each of those assumptions carries meaningful uncertainty, particularly given the regulator’s visible caution in this file following multiple fatalities. The FDA’s decision to request a distribution suspension following the third patient death underscored that the agency is prepared to move decisively on safety signals in this programme.
The broader AAV gene therapy field is watching this safety protocol closely
Sarepta’s approach to managing AAV-associated hepatotoxicity in a high-risk patient population has implications that extend beyond ELEVIDYS and Duchenne. The use of mTOR inhibition as a prophylactic strategy in gene therapy is being evaluated across other programmes, and the data from Cohort 8 will constitute one of the larger prospective datasets examining this approach in a defined clinical setting. If sirolimus prophylaxis proves effective in reducing ALI incidence without compromising transgene expression, the protocol could become a reference point for other AAV gene therapy developers navigating similar hepatic immune challenges, particularly in older or more severely affected patient populations who tend to exhibit stronger pre-existing immune priming.
Roche, which holds the commercial rights to ELEVIDYS outside the United States through its partnership with Sarepta, has imposed its own dosing restrictions in the non-ambulatory population following the fatalities. The international regulatory picture remains distinct from the U.S. pathway, and it is not yet clear whether favourable Cohort 8 data would automatically translate into label restoration in jurisdictions outside the FDA’s remit. The interplay between Sarepta’s U.S.-focused clinical strategy and Roche’s international regulatory obligations adds a layer of complexity to the commercial recovery scenario for this programme. Industry observers note that the scale of Sarepta’s concurrent restructuring, including a 36% workforce reduction and a strategic pivot toward its siRNA platform, makes the ELEVIDYS non-ambulatory question a financially as well as clinically critical variable for the company’s near-term outlook.
What clinicians and regulators will be watching as Cohort 8 progresses
For clinicians managing non-ambulatory patients with Duchenne, the immediate practical question is whether current clinical trial participation through ENDEAVOR Cohort 8 represents the only viable route to access for this population in the near term. The commercial label currently excludes them, and no alternative gene therapies are approved for Duchenne. The unmet need is acute. For regulatory watchers, the test will be whether the FDA treats the Cohort 8 dataset as sufficient to support a pre-market submission or requires a larger, potentially controlled study before reinstating the broader label. The FDA’s willingness to allow Cohort 8 to proceed does not in itself commit the agency to a particular standard of evidence for the subsequent label decision. Primary endpoint data collection is expected in the second half of 2026, setting up a potential pivotal submission window in late 2026 or early 2027.